
Regulación del mercado de medicamentos
El ciclo de vida de un medicamento está conformado por una serie de etapas, que incluyen desde la investigación y desarrollo, el registro sanitario, su comercialización, hasta su eventual retiro del mismo en función de su obsolescencia o reemplazo por tecnologías innovadoras.
CICLO DE VIDA DE LOS MEDICAMENTOS
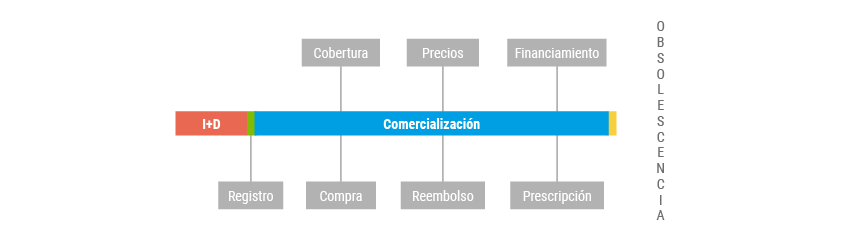
En todas las etapas descriptas es posible implementar estrategias de regulación, pero más allá de las herramientas que podrían implementarse, la segmentación y fragmentación del sistema de salud argentino condicionan que el impacto sobre los diferentes subsectores sea marcadamente desigual.
Hasta el momento, la única barrera que debe atravesar un medicamento para ser comercializado en Argentina es ANMAT (Administración Nacional de Medicamentos, Alimentos y Tecnología Médica), la agencia regulatoria del país. Esta Administración tiene entre sus funciones el registro y autorización, control, fiscalización y vigilancia de la calidad, seguridad y eficacia de los medicamentos, tanto los de síntesis química como los de origen biológico, ingredientes farmacéuticos activos y los excipientes que los componen, para llevar a cabo las acciones de prevención, resguardo y atención de la salud de la población.
Pueden identificarse tres momentos en el desarrollo de las actividades de ANMAT: la habilitación del establecimiento, en el cual se comprueba las instalaciones, equipamiento y servicios destinados a actividades que se utilizan en las fases de elaboración, fraccionamiento, importación y/o exportación, depósito y distribución. Posteriormente, se procede a la fase de registro del medicamento, basados en la verificación de la calidad, seguridad y eficacia de los mismos a través de la evaluación farmacotécnica, pre-clínica y clínica, entre otras. Finalmente, una vez otorgado el registro sanitario, ANMAT cuenta con herramientas para realizar la vigilancia de los productos comercializados durante todo su ciclo de vida.
ACTIVIDADES QUE DESARROLLA ANMAT

Una vez que el medicamento obtiene el registro sanitario y la autorización para su comercialización se encuentra en condiciones de ser prescripto, dispensado, comercializado y financiado por alguno de los actores del sistema de salud.
El mismo año de la creación, se promulgó el Decreto 150/92 donde se establecieron las definiciones de medicamentos y de especialidad medicinal o farmacéutica, las cuales continúan vigentes en la actualidad.
MEDICAMENTOS Y ESPECIALIDAD MEDICINAL O FARMACÉUTICA. DEFINICIONES ANMAT

En ese mismo Decreto se define qué se considera nombre genérico: principio activo o droga farmacéutica o, cuando corresponda, de una asociación o combinación de principios activos a dosis fijas, adoptada por la autoridad sanitaria nacional o, en su defecto, la Denominación Común Internacional de un principio activo recomendada por la Organización Mundial de la Salud; ésta adquiere gran protagonismo al promulgarse la Ley 25.649/02 que promociona la prescripción, dispensación y utilización de medicamentos a través de tal denominación.
Registro de especialidades medicinales o farmacéuticas
La comercialización de especialidades medicinales está sujeta a la aprobación de los productos, previa comercialización, por parte de la Dirección de Evaluación y Registro de Medicamentos (DERM). Esta Dirección, dependiente de INAME, tiene la función de evaluar los trámites de registro de estos productos. Inicialmente, constata que el establecimiento haya obtenido los correspondientes certificados de habilitación emitidos por ANMAT, así como el de Buenas Prácticas de Fabricación (BPF). Posteriormente, la DERM analiza el grado de cumplimiento de los requisitos inherentes a calidad, seguridad y eficacia dispuestos en las normas específicas en las que la ANMAT establece las condiciones para el registro de la amplia diversidad especialidades medicinales o farmacéuticas (en adelante REM, acrónimo de Registro de Especialidades Medicinales).
Como se mencionó previamente, la evaluación de la calidad, seguridad y eficacia de los productos se realiza a partir de información específica aportada por el propio regulado. La información requerida depende del tipo de especialidad medicinal o farmacéutica que se pretenda registrar, el país de origen donde se encuentra la planta de elaboración y de los sitios donde se comercializan los productos.
Los productos a registrar pueden tener origen diverso: medicamentos sintéticos o semisintéticos (que constituyen el grupo con mayor pedido anual de registro) y los de origen biológico. Para ambos casos, ANMAT define requisitos para el análisis de su calidad, seguridad y eficacia que terminarán (o no) en el registro del producto. También establece la información específica que se debe presentar sobre las características del producto a registrar, información de calidad fisicoquímico-farmacéutica y biológica e información preclínica y clínica.
Con el objeto de facilitar la accesibilidad de nuevos agentes terapéuticos, la Administración Nacional creó el Programa de Acceso Expandido (PAE) y estableció, a través de la Disposición 828/1736, los requerimientos para regular medicamentos para grupos de pacientes que requieran tratamientos con productos aún no comercializados en el país, destinados al tratamiento de patologías graves y que podrían haber obtenido recientemente las evidencias clínicas experimentales suficientes como para iniciar el proceso de registro y autorización de comercialización ante otra agencia regulatoria perteneciente al Anexo I del Decreto 150/92.
Sistemas de vigilancia
Una vez que un medicamento recibe la autorización para su comercialización, se inicia la fase de vigilancia del producto, actividad que es llevada a cabo por la ANMAT y el laboratorio farmacéutico en forma conjunta. Esta fase tiene por finalidad monitorear, tanto en el ámbito nacional como internacional, su seguridad y eficacia.
A través de su Sistema Nacional de Farmacovigilancia (SNFV), creado por la Resolución 706/93 del entonces Ministerio de Salud y Acción Social, ANMAT ejerce sus acciones de control y fiscalización de las especialidades medicinales, con el fin de detectar precozmente: reacciones adversas graves, sospecha de reacciones adversas durante los primeros 5 años, interacciones con otros medicamentos, alcohol y alimentos, percepción de fallas a respuestas terapéuticas.
Para contribuir a erradicar la circulación de productos ilegítimos, ANMAT creó el Sistema Nacional de Trazabilidad de Medicamentos diseñado para que, de manera eficiente y en tiempo real, se trace el recorrido de cada medicamento desde su elaboración hasta su dispensación. Este sistema es implementado por toda persona física o jurídica que interviene en la cadena de comercialización, distribución y dispensa de especialidades medicinales (laboratorios, distribuidoras, operadores logísticos, droguerías, farmacias, establecimientos asistenciales, establecimientos estatales, botiquines de farmacia y laboratorios elaboradores de soluciones nutricionales de uso inmediato). A partir de la creación del Sistema de Trazabilidad, quienes quieran comercializar, distribuir y dispensar productos deben asociarlo al código unívoco, datos de la distribución denominados código del destinatario (Global Location Number (GLN), por sus siglas en inglés), fecha en que se realizó la transacción y la factura y/o remito asociado a la operación de distribución o dispensa.
Sumado a las actividades mencionadas, ANMAT cuenta con el Programa de Evaluación de Tecnologías Sanitarias (PETS), que evalúa la efectividad de las tecnologías sanitarias autorizadas y comercializadas en el mercado local. El objetivo del PETS es proveer de informes a la Administración Nacional para la toma de decisiones, basados en la mejor evidencia disponible, sobre vigencia terapéutica, usos novedosos de productos registrados o sobre aquellas tecnologías en fases muy tempranas de investigación.